
In today’s biopharma landscape, microbial versatility isn’t a luxury—it’s a necessity. For decades, Escherichia coli has been the microbial workhorse of the CDMO world, offering fast growth, simple genetics, and ease of scale-up. But the biologics being made today—complex enzymes, glycosylated proteins, biosimilars, and new modalities—are pushing the limits of what E. coli can deliver. More than ever, CDMOs are being asked to evolve: to embrace organisms like Pichia pastoris, Saccharomyces cerevisiae, and filamentous fungi such as Aspergillus niger or Trichoderma reesei.
These hosts introduce new possibilities—secretion, folding, and glycosylation—but also new challenges. Introducing a new microbial strain isn’t a plug-and-play switch. It’s a comprehensive transformation across feasibility, process development, analytics, regulatory, and operational dimensions. Below is your detailed roadmap for doing it right.
🔬 1. Strain Evaluation & Feasibility
Redefining the Foundation: A Scientific and Strategic Choice
The decision to adopt a new microbial strain is one of the most consequential steps in CDMO operations. It defines not only your production capabilities but also the technical boundaries of what kinds of molecules you can manufacture—now and into the future. As therapeutic pipelines grow more complex—with rising interest in glycoproteins, enzymes, and metabolic pathway engineering—traditional systems like E. coli begin to show their limits.
For CDMOs, this isn’t just a scientific consideration—it’s a business-critical decision. Clients expect speed, scalability, and flexibility. Switching to a eukaryotic system like Pichia pastoris, Saccharomyces cerevisiae, or even filamentous fungi can unlock expression capabilities inaccessible to prokaryotic platforms. But it also means greater technical risk. Before any bioreactor is fired up or plasmid is transformed, CDMOs must conduct rigorous feasibility and strain evaluation studies that account for biological performance, regulatory compliance, scalability, and commercial viability.
A robust strain evaluation framework addresses these risks up front—ensuring that downstream investment in process development, analytics, and tech transfer are built on solid ground.
📌 Deep-Dive Feasibility Criteria:
1. Genetic Stability & Expression Architecture
- Assess whether the plasmid or chromosomal integration remains stable through at least 10–15 passages under representative selection pressure.
- Evaluate copy number variation (especially in Pichia) and its impact on expression—too many copies can burden the host and induce proteotoxic stress.
- For inducible systems (e.g., AOX1 in Pichia), test promoter leakiness and repression efficiency under carbon source shifts.
- Analyze codon optimization of the gene of interest (GOI) for host compatibility—rare codons may stall translation and reduce titer.
- Perform Southern blot or qPCR to confirm integration locus and copy number homogeneity in stable lines.
2. Host Growth Kinetics & Bioprocess Compatibility
- Measure lag phase duration, exponential growth rate (µ), and maximum OD600 or dry cell weight (DCW) achievable under defined media.
- Optimize temperature, pH, and agitation setpoints for both shake flask and bioreactor formats—E. coli may prefer 37°C, while Pichia thrives at 30°C.
- Conduct carbon source utilization profiling (e.g., glycerol, glucose, methanol, sorbitol) to plan feed strategies.
- Determine oxygen uptake rate (OUR), carbon dioxide evolution rate (CER), and specific growth rate (SGR) under controlled bioreactor conditions to build a kinetic model.
- Evaluate oxygen demand using respirometry or DO spike testing to pre-model oxygen transfer limitations in larger tanks.
3. Protein Folding, Secretion, and PTM Support
- Use small-scale expression to test protein solubility, secretion efficiency, and proper folding—especially for proteins with multiple disulfide bonds or complex tertiary structures.
- Run SDS-PAGE with and without reducing agent to check disulfide bridge formation.
- Conduct glycosylation profiling (e.g., HILIC-UPLC or LC-MS) for eukaryotic strains—verify whether the glycoforms are high-mannose, hybrid, or complex types.
- Determine the strain’s ability to secrete product into the supernatant. For Pichia, the α-mating factor signal peptide is commonly used—test cleavage efficiency and impact on final yield.
- For enzymes, test co-factor incorporation or metal ion dependency (e.g., Zn²⁺, Mn²⁺)—host metal homeostasis can impact catalytic activity.
4. Product Compatibility & Manufacturability
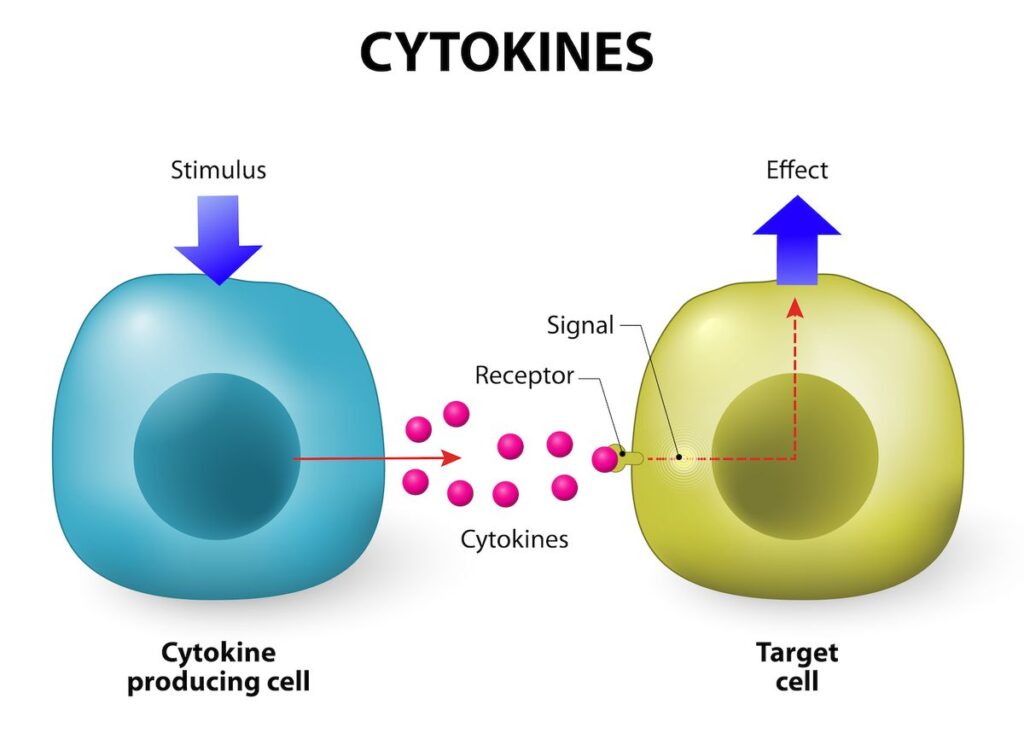
- Evaluate whether the strain is capable of expressing membrane-bound proteins, large multi-domain proteins, or high-molecular-weight complexes.
- Run thermal shift assays or DSF (Differential Scanning Fluorimetry) to assess product stability in crude lysate or secreted form.
- For fusion proteins or antibody fragments, test linker stability, aggregation propensity, and functional binding via ELISA or SPR.
- Consider the presence of host proteases—some fungi and yeasts secrete proteases under stress or stationary phase, which can degrade your product unless inhibited or genetically knocked out.
5. Scalability & Economic Considerations
- Model media cost per liter and cost per gram of product at target yields—include methanol, glycerol, nitrogen, vitamins, and trace minerals.
- Estimate biomass generation and total broth volume needed to meet clinical batch demand.
- Simulate oxygen transfer rate (OTR) and kLa at various scale heights—critical for high-density cultures such as those in Pichia.
- Evaluate the host’s safety profile and regulatory history—Pichia pastoris and Saccharomyces cerevisiae are generally recognized as safe (GRAS), while filamentous fungi may require more rigorous toxicology.
🧪 Real-World Case Example:
A CDMO was developing a glycosylated cytokine that failed to express in E. coli without forming inclusion bodies. They shifted to Pichia pastoris, leveraging the AOX1 promoter system with α-factor secretion. While initial expression levels were 40% lower, the bioactivity doubled due to proper folding and glycosylation. This eliminated the need for refolding and reduced chromatography burden downstream, ultimately lowering cost of goods (COGS) and improving timeline to GMP production.
⚙️ 2. Upstream Process Development
Engineering the Right Environment: Where Biology Meets Bioreactor
Once a strain passes feasibility screening, the real work begins—optimizing the environment in which it will grow, express, and thrive. Upstream process development is more than just dialing in growth conditions; it’s about designing a biological ecosystem that reliably pushes the organism to perform at its best, batch after batch.
The transition from E. coli to more complex microbes like yeast or fungi shifts the paradigm. E. coli is fast, predictable, and largely forgiving. In contrast, organisms like Pichia pastoris or Aspergillus niger demand greater precision in carbon source control, oxygen management, and phase transitions. These hosts offer powerful benefits—such as secretion and proper folding—but they make upstream development a much more nuanced and interdisciplinary effort.
What works in a shake flask won’t always work at 200L, and what works at 200L might not behave the same at 2,000L. That’s why this phase must be rigorous, iterative, and grounded in an understanding of both biology and engineering.
🔍 Core Development Areas:
Media Optimization
- Tailor base media to host-specific carbon and nitrogen needs. Pichia often prefers glycerol for biomass, methanol for induction—both must be fed gradually to avoid toxicity or repression.
- Design feed strategies using carbon-limited exponential profiles or DO-stat control to maintain consistent growth without overflow metabolism.
- Minimize accumulation of byproducts like ethanol (in yeast) or acetate (in E. coli) by controlling dissolved oxygen and feed concentration.
- Consider adding vitamins (biotin, thiamine), trace metals (Zn, Cu, Fe), and pH stabilizers for high-density fermentation performance.
Fermentation Strategy
- Choose mode based on product and host: batch for simplicity, fed-batch for titer maximization, and continuous for long campaigns or high-throughput production.
- Pichia fed-batch systems often use a multi-phase strategy: glycerol batch → glycerol fed-batch → methanol induction.
- In fungi, monitor broth viscosity and morphology: mycelial growth can cause mixing and aeration issues, while pellet formation may improve processability.
Critical Process Parameters
- Precisely control DO (% air saturation), temperature, agitation, and pH using PID loops tuned to the strain’s specific respiratory demand.
- Oxygen uptake rate (OUR) and carbon dioxide evolution rate (CER) should be monitored in real-time to predict phase transitions and induction points.
- Evaluate shear sensitivity—some fungi suffer from cell wall lysis under high agitation, requiring low-shear impeller design or modified spargers.
Scale Translation
- Model kLa, mixing time, and CO₂ stripping efficiency at pilot scale to inform scale-up factors.
- Address foam generation early—especially in yeast or fungal fermentations. Validate antifoam agents for biocompatibility and downstream clearance.
- Anticipate pressure drops in filters and probes clogged by fungal clumps; inline screening or vortex-based sensors may be required.
🧪 Real-World Insight:
A CDMO scaling Aspergillus niger for enzyme production encountered unexpected broth thickening due to pellet fragmentation. The oxygen transfer rate (OTR) dropped below viability threshold. The solution: replacing the Rushton impeller with a marine-style impeller and integrating microbubble aeration. This not only restored viability but increased enzyme productivity by 20%.
🧪 3. Downstream Process Development
Purifying Complexity: Adapting Recovery to a New Biological Signature
Once a new microbial strain is chosen and upstream parameters are locked in, the next challenge is just as critical—downstream processing. This is where the biology of the organism meets the realities of chemistry, filtration, and separation. While E. coli often requires brute-force extraction and elaborate refolding strategies, hosts like yeast or fungi introduce different hurdles: secreted products, higher molecular weights, glycosylation, and unfamiliar impurity profiles.
The shift from intracellular recovery to extracellular secretion can dramatically reduce processing steps—but it also demands an entirely different playbook. Filtration, clarification, chromatography, and buffer systems must all be reassessed to match the expression environment, the nature of the host cell components, and the stability of the target molecule.
Here, the goal isn’t just purity—it’s processability, yield retention, and maintaining product integrity throughout multiple unit operations. A small oversight in early clarification can clog filters downstream; an unanticipated glycan structure can wreak havoc on an affinity column. Every decision is connected.
🧬 Key Areas of Adaptation:
Lysis vs. Secretion: A Paradigm Shift
- E. coli products are often trapped in inclusion bodies, requiring high-pressure homogenization or sonication—followed by solubilization and refolding.
- Yeasts and fungi frequently secrete proteins directly into the culture medium, bypassing lysis and reducing biomass removal burden.
- Secretion streamlines clarification but introduces host-derived secreted proteases and polysaccharides that must be managed downstream.
Clarification & Filtration
- For secreted proteins, use depth filters and centrifugation to remove cells and cell debris without shearing.
- Fungal broths often require pre-filtration or flocculants to reduce viscosity before depth filtration.
- Validate filter compatibility for fouling resistance—beta-glucans and mannoproteins in yeast can clog standard TFF or sterile filters.
Impurity Profile Redesign
- Endotoxins (a major concern in E. coli) are largely absent in yeast/fungi but replaced by:
- Mannans
- Glucans
- Chitin fragments
- Proteolytic degradation products
- These impurities may not trigger the same innate immune responses but still affect purity, stability, and regulatory compliance.
Purification Scheme Optimization
- Reoptimize IEX, HIC, and affinity protocols to match product charge, hydrophobicity, and glycosylation status.
- Glycoproteins may interact unpredictably with resin matrices—sometimes requiring deglycosylation or use of lectin-affinity steps.
- Elution buffers may need modification (e.g., arginine-based formulations) to prevent aggregation or improve solubility for heavily glycosylated proteins.
Stability and Aggregation Risks
- Glycosylation may stabilize the protein, but also lead to charge heterogeneity. Monitor using isoelectric focusing or cIEF.
- Analyze aggregation using SEC-MALS or DLS to avoid downstream filter fouling or chromatographic losses.
- Host proteases secreted during late-log or stationary phase can degrade the product—add protease inhibitors or design harvest triggers accordingly.
🧪 Real-World Example:
In a CDMO project involving Trichoderma reesei, the client’s cellulase product was secreted in large amounts—but polysaccharide contaminants began fouling downstream TFF units. A redesign implemented inline prefiltration, pH-controlled tangential flow loops, and an anti-fouling filter coating. This not only reduced downtime by 40% but maintained product yield and cut COGS across five campaigns.
🧬 4. Analytical Development
Redefining Quality: When New Biology Requires New Eyes
Switching microbial strains doesn’t just change what you’re growing—it changes how you measure, qualify, and release the final product. Analytical development is where the true complexity of strain switching often reveals itself. Glycosylation, host cell impurities, folding states, charge heterogeneity—these aren’t just biological curiosities; they’re critical quality attributes (CQAs) that define product identity, safety, and efficacy.
A method validated on E. coli-derived product can fail spectacularly when applied to Pichia, Saccharomyces, or Aspergillus. The host background changes everything: the HCP profiles, nucleic acid contaminants, glycan structures, secretion signatures, and even matrix effects in ELISA or qPCR. You’re not just rechecking old boxes—you’re redrawing the entire map of how your product is evaluated.
Analytical readiness must be part of the process from the first feasibility screen, not an afterthought before IND filing. It informs comparability, supports regulatory strategy, and ensures that the process you’ve built delivers a molecule that behaves the way the patient—and the agency—expects.
🧪 Development Priorities:
Assay Re-Validation for New Host Backgrounds
- ELISA assays for host cell proteins (HCPs) are host-specific. An HCP ELISA for E. coli will miss Pichia or fungal impurities entirely—develop or source strain-specific polyclonal antibodies.
- qPCR primers for residual DNA quantification must be redesigned based on the new host genome—S. cerevisiae and Pichia have vastly different GC content and repetitive elements.
- SDS-PAGE and Western blots may reveal unexpected bands due to glycosylation or secretion signal remnants—re-validate molecular weight ladders and detection antibodies.
- If using product-specific binding assays (e.g., receptor-ligand), test binding kinetics pre- and post-glycosylation shift using SPR (e.g., Biacore) or BLI.
Product Characterization and Structural Integrity
- Use LC-MS or MALDI-TOF to determine exact mass and glycoform distribution. Glycosylation from yeast often results in high-mannose or hyperglycosylated variants, which may differ from human-like patterns.
- Run capillary isoelectric focusing (cIEF) or imaged cIEF to resolve charge variants introduced by glycosylation or misprocessing.
- Evaluate protein folding via circular dichroism (CD), thermal shift assays (DSF), or fluorescence-based stability screens.
- For fusion proteins or antibody fragments, assess linker integrity and domain folding by SEC-MALS or peptide mapping.
Functionality and Bioassay Development
- Use cell-based assays to test receptor binding, downstream signaling, or enzymatic activity. Glycosylation can enhance or inhibit these interactions—especially in cytokines, hormones, or Fc-fusion proteins.
- Compare potency curves between old and new strains during early process development. A change in EC50 may reflect folding differences or altered glycoform interactions.
- Incorporate accelerated stability and forced degradation studies to evaluate how strain-related modifications affect long-term storage behavior.
Host-Specific Impurity Analysis
- Characterize beta-glucans, mannoproteins, and chitin in yeast and fungi. These are not typically immunogenic in humans but may impact formulation, filter performance, or viscosity.
- Use DNA laddering assays or qPCR to quantify residual host DNA; define acceptable thresholds per ICH Q6B.
- Monitor protease activity in supernatants, particularly during stationary phase—develop protease activity assays and spike-in controls during purification validation.
🧪 Real-World Insight:
A CDMO transitioning a recombinant vaccine antigen from E. coli to Pichia noticed a consistent shift in SDS-PAGE mobility. Initial concerns pointed to product truncation, but intact mass analysis via LC-MS revealed extensive glycosylation. Surprisingly, this altered glycoform conferred improved thermal stability and better antigen presentation in preclinical studies—data that later strengthened the client’s regulatory submission and supported differentiated product claims.
🏗️ 5. Tech Transfer & Scale-Up
Bringing Biology into Steel: Where Precision Meets Pressure
After the strain is optimized and lab-scale processes are developed, the real test begins: translating biology into equipment. Tech transfer is the crucible where upstream and downstream theory meets the cold, hard steel of real-world manufacturing. At this stage, variability is magnified. Minor oversights can become batch failures. And process control isn’t just about science—it’s about engineering repeatability, equipment fit, facility capability, and cross-functional fluency.
For CDMOs expanding beyond E. coli, the stakes are even higher. New strains often behave unpredictably at scale: Pichia might foam excessively during methanol induction; Aspergillus can create viscous broths that defy oxygen transfer and sensor reliability. Equipment that once worked flawlessly with bacterial fermentations may now underperform—or actively disrupt—the performance of yeast or fungal processes.
A successful tech transfer involves more than passing a protocol. It requires building a robust, data-backed bridge from bench to plant. That means scaling not just the biology but also the controls, cleaning strategies, documentation, and even the assumptions behind the process itself.
🧩 Core Components of a Successful Tech Transfer:
Comprehensive Documentation & Knowledge Capture
- Develop a full Process Transfer Package (PTP), including:
- Process flow diagrams (PFDs)
- Raw material specifications with alternate vendor options
- Defined ranges for critical process parameters (CPPs) and in-process controls (IPCs)
- Include microbial strain history, vector maps, passage numbers, and induction kinetics as part of upstream records.
- Document equipment compatibility assumptions—e.g., maximum agitation rates, sparger design, pump pressure tolerances.
Pilot Studies & Engineering Runs
- Perform 10–100L engineering batches in the receiving site’s vessels to model foam generation, oxygen demand, and mixing profiles.
- Monitor dynamic process variables like DO spikes, pH drifts, or off-gas CO₂ to anticipate control loop lags or PID tuning needs at production scale.
- Capture real-time data using process analytical technology (PAT) to correlate scale-dependent behaviors with lab data.
Equipment Compatibility Assessment
- Validate that fermenters can maintain oxygen transfer rates (kLa > 180 h⁻¹ for Pichia) and handle the gas flow required for methanol metabolism or mycelial oxygen demand.
- Check if impeller design supports proper mixing at high broth viscosities—marine or pitched-blade impellers may replace Rushton turbines for fungal cultures.
- Confirm filtration and chromatography equipment can handle host-specific impurities and product loads at scale—especially if secreted products are present in high concentrations.
Control Strategy Translation
- Translate lab-based process triggers (e.g., OD600, DO drop, pH inflection) into scalable, equipment-driven control logic.
- Validate soft sensors or automation platforms (e.g., methanol feed algorithms, foam sensors, or real-time oxygen controllers) under manufacturing-scale stress conditions.
- Implement alarm limits, failsafe shutoff routines, and escalation procedures aligned with microbial metabolism and toxicity risks (e.g., methanol overfeeding).
🧪 Real-World Example:
A CDMO scaling Saccharomyces cerevisiae to a 2,000L fermenter underestimated the foaming response to aggressive aeration during induction. Within 90 minutes of methanol feed initiation, foam overflowed into headspace filters and triggered a plant-wide shutdown. Postmortem analysis revealed that legacy defoaming protocols were tuned for E. coli—not yeast. The team responded by validating low-toxicity antifoam agents, redesigning baffles, and installing mechanical foam breakers.
This event delayed the campaign by two weeks—but the retrofits became a permanent infrastructure upgrade, enabling future fungal programs to run safely at scale.
🧻 6. Regulatory & Quality Considerations
Translating Innovation into Compliance: Speaking the Regulator’s Language
Adopting a new microbial host isn’t just a scientific leap—it’s a regulatory one. Even if the biology works and the manufacturing flows, none of it matters if your documentation, comparability studies, and impurity data don’t align with current expectations from agencies like the FDA or EMA. Here, biology must become narrative: data becomes story, and your job is to prove that the new system is not just equivalent—but justifiably better, safer, or more robust.
While E. coli systems have a long-established regulatory framework, alternative hosts—especially yeasts and fungi—bring additional scrutiny. Are the impurities known? Is the glycosylation safe? Can you prove consistency across batches, scales, and tech transfers? For CDMOs, the key is to be proactive, not reactive—to anticipate questions before they’re asked, and to frame the transition as a risk-managed enhancement of capability.
🧾 Key Regulatory Priorities:
Host Safety and Regulatory Precedence
- Confirm the host strain has GRAS status or has been used in previous biologics with regulatory approval.
- For non-GRAS organisms or modified strains (e.g., protease knockouts), compile toxicology data or support literature demonstrating safety and precedent.
GMP Documentation Revisions
- Update the Master Batch Record (MBR), Bill of Materials (BOM), and all SOPs to reflect the new process and host.
- Revise environmental monitoring (EM) plans to account for spores or unique metabolites—especially important for fungi like Aspergillus.
- Reassess cleaning validation and shared-equipment matrix: beta-glucans or fungal debris may require different detergents or contact times.
Change Control & Comparability Strategy
- Conduct full comparability assessments per ICH Q5E if switching hosts mid-development or for biosimilar production.
- Use orthogonal methods (e.g., LC-MS, SPR, bioassays) to show that CQAs—such as glycosylation, potency, and aggregation—are equivalent or superior.
- Include stability data and forced degradation profiles to demonstrate that the new expression system maintains shelf-life and formulation integrity.
🧪 Real-World Example:
When a CDMO transitioned a biosimilar cytokine from E. coli to Pichia, they faced agency concerns about altered glycan profiles. While structurally different, the new glycoforms showed enhanced serum stability and potency in vitro. The team presented this as a functional improvement, supported by pharmacodynamic data and receptor-binding affinity assays. The amendment to the IND was approved with no delay.
🧠 7. Training & Facility Readiness
Aligning the Human System with the Biological One
Even the best process will collapse without operational alignment. When a new strain enters the plant, it doesn’t just change the SOPs—it changes how your people think, how they clean, how they gown, and how they respond when things go off-script. The entire facility becomes a learning organism—and the strain becomes a stress test for both systems and culture.
The biological risks are real: fungal spores escaping containment, unknown proteases interfering with product integrity, unfamiliar QC signals being misread. And the only way to mitigate these is with structured retraining, proactive cross-functional alignment, and a willingness to reimagine old assumptions.
🏭 Operational Adjustments:
Staff Retraining & Process Awareness
- Upstream teams must learn new fermentation kinetics, induction triggers, and foam control protocols.
- Downstream operators need hands-on training with secretion-based harvests, filtration challenges, and new chromatography strategies.
- QA/QC personnel must be trained on new impurity profiles, strain-specific ELISA interpretations, and abnormal trend detection.
Cleanroom Practices & Facility SOPs
- Review gowning and aseptic handling for spore-forming organisms—especially in Grade C or D environments.
- Upgrade air handling and HEPA filtration if spore or aerosol control is at risk; conduct airflow smoke studies to confirm containment.
- Adjust cleaning regimens for fungal debris or yeast residues—use sporicidal agents like hydrogen peroxide or peracetic acid.
Safety & Biosafety Protocols
- Conduct a biosafety assessment for each strain—consider allergenic potential, pathogenicity, and potential environmental impact.
- For certain fungi, implement additional spore traps or settle plates near entry/exit points.
🧪 Real-World Example:
A CDMO incorporating Aspergillus niger discovered spores in Grade C areas during routine EM—despite standard GMP protocols. Root cause analysis revealed lapses during vial transfers and open-hatch centrifuge loading. The team revised gowning SOPs, introduced peroxide wipe stations, and updated EM frequency. Contamination levels dropped by 95% in one quarter.
🧬 What the Microbe Teaches Us
Listening to the Cell, Evolving the System
Switching microbial strains is more than a technical process—it’s a transformation of mindset. It asks a CDMO not just to solve problems, but to ask better questions:
What does the cell need? What are we missing in our current approach? How can our systems become as adaptive as the biology we work with?
E. coli taught us how to scale biology.
Yeast teaches us how to fold it, secrete it, and refine it.
Fungi challenge us to stretch the limits of oxygen, viscosity, and operational control.
But beneath all these differences lies a deeper truth: the cell is alive. It resists simplification. It evolves. And in learning to respect that, we evolve with it.
A CDMO that can seamlessly shift between microbial dialects doesn’t just gain technical range—it becomes a partner that biotech innovators can trust with their most ambitious projects. In this complexity, we find new opportunity:
- Opportunity to offer clients broader expression platforms.
- Opportunity to rescue difficult-to-express targets.
- Opportunity to pioneer solutions where others default to “can’t.”
Biotech is a language. Each microbe is a different way to say the same thing: create something that heals. And those who learn to speak more languages will be the ones who shape the future.
Let the microbe teach you. Let it challenge your system. And in doing so, let it refine not just your process—but your entire way of building medicine!
